
Sickle Cell
What is Sickle cell?
The term sickle cell disease (SCD) describes a group of inherited red blood cell disorders. People with SCD have abnormal hemoglobin, called hemoglobin S or sickle hemoglobin, in their red blood cells.
Hemoglobin is a protein in red blood cells that carries oxygen throughout the body.
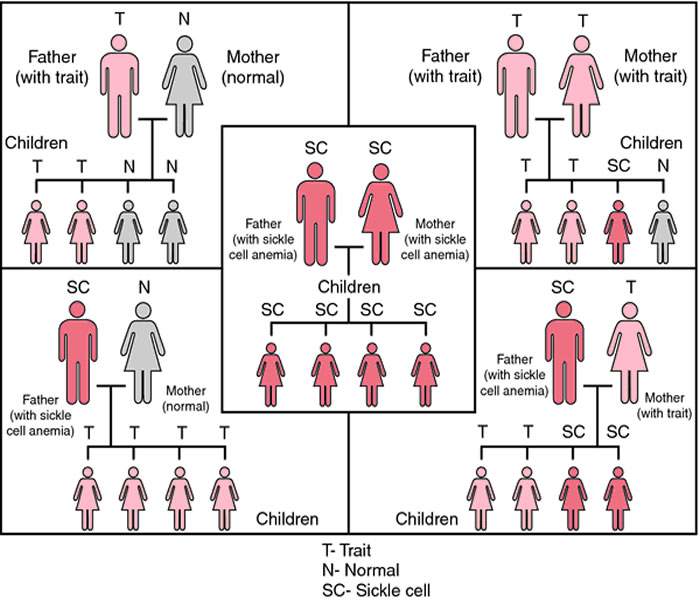
“Inherited” means that the disease is passed by genes from parents to their children. SCD is not contagious. A person cannot catch it, like a cold or infection, from someone else.
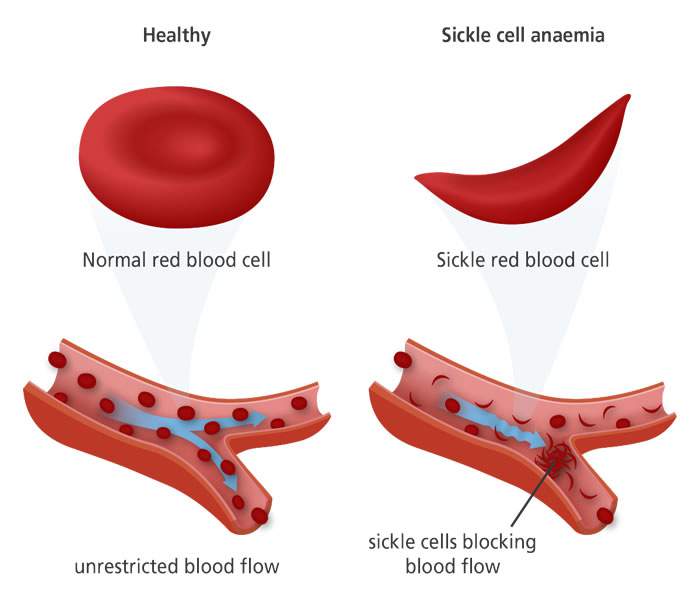
Sickle cell disease is a group of disorders that affects hemoglobin, the molecule in red blood cells that delivers oxygen to cells throughout the body. People with this disorder have atypical hemoglobin molecules called hemoglobin S, which can distort red blood cells into a sickle, or crescent, shape.
Causes of Sickle cell
Sickle cell disease is caused by a faulty gene that affects how red blood cells develop.
If both parents have this faulty gene, there's a 25% chance of each child they have being born with sickle cell disease.
Sickle cell disease is caused by a genetic abnormality in the gene for hemoglobin, which results in the production of sickle hemoglobin. When oxygen is released from sickle hemoglobin, it sticks together and forms long rods, which damage and change the shape of the red blood cell. The sickle red blood cells causes the symptoms of sickle cell disease.
Children are born with sickle cell disease; it is not contagious. It occurs when a child inherits two sickle hemoglobin genes, one from each parent.People who inherit only one sickle hemoglobin gene are carriers (sickle cell trait) and do not have anemia or painful sickle cell crises. They may, though, have a slightly higher incidence of certain conditions such as blood in the urine or urinary tract infections.
Red blood cells deliver oxygen to working or active tissues. In the lungs, hemoglobin (the molecule in the red blood cell) takes on oxygen and, at the same time, releases carbon dioxide. This process is called oxygenation. At the tissue level, this activity is reversed. The same hemoglobin molecule releases oxygen and takes on carbon dioxide. This process is called deoxygenation.
In sickle cell disease, certain red blood cells become crescent-shaped (the sickle cell Dr. Herrick described).
These abnormal red blood cells, carrying abnormal hemoglobin known as hemoglobin S, are fragile.
A person who has sickle cell disease can become more likely to get infections because the damaged cells eventually clog the spleen.
A severe attack, known as sickle cell crisis, can cause pain because blood vessels can become blocked or the defective red blood cells can damage organs in the body.
There is also an impairment in oxygentation from the abnormal hemoglobin S.
How Is Sickle Cell Disease Inherited?
When the hemoglobin S gene is inherited from only one parent and a normal hemoglobin gene is inherited from the other, a person will have sickle cell trait. People with sickle cell trait are generally healthy.
Only rarely do people with sickle cell trait have complications similar to those seen in people with SCD. But people with sickle cell trait are carriers of a defective hemoglobin S gene. So, they can pass it on when they have a child.
If the child’s other parent also has sickle cell trait or another abnormal hemoglobin gene (like thalassemia, hemoglobin C, hemoglobin D, hemoglobin E), that child has a chance of having SCD.
Symptoms/Signs of Sickle cell
Early Signs and Symptoms
If a person has sickle cell disease (SCD), it is present at birth. But most infants do not have any problems from the disease until they are about 5 or 6 months of age. Every state in the United States, the District of Columbia, and the U.S. territories requires that all newborn babies receive screening for SCD. When a child has SCD, parents are notified before the child has symptoms.
Some children with SCD will start to have problems early on, and some later. Early symptoms of SCD may include:
- Anemia
- Arthritis
- Bacterial Infections
- Dactylitis (swelling and inflammation of the hands and/or feet)
- Eye Damage
- Fatigue
Other problems/Advance symptoms
Sickle cell disease can also sometimes cause a wide range of other problems, including:
- Acute Chest Syndrome, which can cause a fever, cough, chest pain, and breathing difficulties
- Anemia
- Aseptic Necrosis and Bone Infarcts (death of portions of bone)
- A yellowish color of the skin, known as jaundice, or whites of the eyes, known as icteris
- Bone and joint pain
- Delayed growth during childhood and delayed puberty
- Gallstones (small stones in the gallbladder), which can cause tummy (abdominal) pain and yellow skin and eyes (jaundice)
- Hand-Foot Syndrome
- Infections
- Kidney or urinary problems, including blood in the urine and bedwetting
- Leg ulcers (painful, open sores on the lower legs)
- Liver Congestion
- Pain Episodes
- Strokes or transient ischaemic attacks (where the flow of blood to the brain is blocked or interrupted)
- Priapism (a persistent and painful erection of the penis), which can sometimes last several hours
- Pulmonary hypertension (high blood pressure in the blood vessels that carry blood from the heart to the lungs)
- Splenic Sequestration (sudden pooling of blood in the spleen)
- Swelling of the spleen, which can cause shortness of breath, a rapid heartbeat, abdominal pain, a swollen tummy and worse anaemia
- Tissue Damage
- Vision problems, such as floaters, blurred or patchy vision, reduced night vision and occasionally sudden vision loss
Acute Chest Syndrome: This is a potentially life-threatening condition that should be treated in a hospital. Caused by an infection or blocked blood vessels in the lung, the condition is similar to pneumonia.
Anemia: Red blood cells containing hemoglobin S do not live as long as normal red blood cells, resulting in a low blood count, a condition called anemia.
Delayed growth: Anemia may cause a child to have slow growth, delayed puberty, pale complexion, tire easily and experience shortness of breath.
Hand-Foot Syndrome: Often the first symptom in affected babies, occurring with a fever, hands and feet swell when blood vessels become blocked by sickle cells.
Infections: Infants and children with sickle cell disease are especially prone to bacterial infections, such as those that cause meningitis and blood infections. Such infections are a leading cause of death in infants and young children with sickle cell disease. They can be prevented by immunization and prophylactic penicillin.
Pain Episodes: the most common symptom of sickle cell disease, pain episodes occur in any place in the body where sickle cells collect and block blood vessels. Episodes vary in length and frequency for each person.
Stroke: An estimated 10 percent of children with sickle cell anemia develop symptoms of a stroke, which occurs when a blood vessel in the brain becomes blocked, and another 20 percent are found to have clinically silent strokes by MRI imaging of their brains. Stroke may result in permanent disability or learning problems. However, doctors are now able to identify most children who are at an increased risk of having a symptomatic stroke with a special ultrasound exam.
Tissue Damage: When sickle cells block small blood vessels, the amount of blood received by certain parts of the body is reduced. Tissue that does not receive an adequate amount of blood will eventually become damaged.
Vision Problems: Vision problems and in some cases, blindness, may occur when blood vessels in the eye become blocked by sickle cells. by ucsfbenioffchildrens.
Some features of sickle cell anemia, such as fatigue, anemia, pain crises, and bone infarcts can occur at any age. Many features typically occur in certain age groups.
Sickle cell anemia usually first presents in the first year of life. Infants and younger children can suffer with fever, abdominal pain, pneumococcal bacterial infections, painful swellings of the hands and feet (dactylitis), and splenic sequestration. Adolescents and young adults more commonly develop leg ulcers, aseptic necrosis, and eye damage. Symptoms in adult typically are intermittent pain episodes due to injury of bone, muscle, or internal organs.
Affected infants do not develop symptoms in the first few months of life because the hemoglobin produced by the developing fetus (fetal hemoglobin) protects the red blood cells from sickling. This fetal hemoglobin is absent in the red blood cells that are produced after birth so that by 5 months of age, the sickling of the red blood cells is prominent and symptoms begin.
How to diagnose Sickle cell
Your doctor will give you a physical exam and may ask you questions like:
Where do you usually feel pain?
How frequently do you get it?
How long does it last?
Do you get a lot of infections?
Do you often feel tired?
Your doctor will also ask you to get your blood tested to see if it has sickle cell hemoglobin, the faulty protein that causes your red blood cells to have that shape.
The test uses blood from the blood samples used for other routine newborn screening tests. It can show whether a newborn infant has sickle cell disease or sickle cell trait. If the test shows sickle hemoglobin, a second blood test is done to confirm the diagnosis.
It's also possible for doctors to diagnose sickle cell disease before birth. This is done using a sample of amniotic fluid or tissue taken from the placenta. (Amniotic fluid is the fluid in the sac surrounding a growing embryo. The placenta is the organ that attaches the umbilical cord to the mother's womb.) This test can be done in the first few weeks of pregnancy.
What does the test result mean?
Newborn screening
In newborns who carry the sickle cell gene, fetal hemoglobin F will predominate but a small amount of hemoglobin S will also be present. There may be a small amount of hemoglobin A if they have sickle cell trait. A full work-up should be done after the child reaches six months of age.
Diagnostic testing
Adults with sickle cell trait will produce mostly normal hemoglobin A, while those with sickle cell disease (anemia) will produce mostly Hb S with no Hb A. People who are heterozygous for two different hemoglobin variants will usually produce varying amounts of both types. For example, they may produce both Hb S and Hb C but no Hb A.
Genetic testing
If two copies of the Hb S gene mutation are detected, then the person has sickle cell disease. If the person has one gene that codes for Hb S and one normal gene, then the person has sickle cell trait. If the person has one Hb S copy and a Hb C or beta thalassemia mutation, then the person is likely to experience some symptoms and complications associated with sickle cell disease. If the person has one Hb S gene copy and another, more rare hemoglobin variant, then the person may or may not have any symptoms or complications. See the article on Hemoglobin Abnormalities for more on this.
What is a sickle cell crisis?
A sickle cell crisis is pain that can begin suddenly and last several hours to several days. It happens when sickled red blood cells block small blood vessels that carry blood to your bones. You might have pain in your back, knees, legs, arms, chest or stomach. The pain can be throbbing, sharp, dull or stabbing. How often and how bad the pain gets varies a lot from person to person and from crisis to crisis.
You might be able to treat your pain crisis at home with medicines that you take by mouth. If these medicines don't control your pain, you can't keep fluids down or you know that you're having severe pain, you might need to be treated in the emergency department. If your pain still isn't controlled or you have other problems, you might need to be treated in the hospital.
How to prevent a sickle cell crisis?
- Ask about vaccinations you need.
- Balance rest and exercise.
- Do not smoke cigarettes or drink alcohol.
- Drink liquids as directed.
- Stay out of the cold.
- Take vitamins and minerals as directed. Folic acid can help prevent blood vessel problems that can come with sickle cell anemia.
How to Prevent Sickle cell
Sickle cell disease is an inherited blood disorder that is not preventable.
People who do not know whether they carry an abnormal hemoglobin gene can ask their doctor to have their blood tested.
Couples who are planning to have children and know that they are at risk of having a child with sickle cell disease (SCD) may want to meet with a genetic counselor.
If you carry the sickle cell trait, meet with a genetic counselor before trying to conceive a child. A genetic counselor can help you understand your risk of having a child with sickle cell anemia. If you are a carrier (AS genotype) do not marry a carrier (AS genotype) if you want have child or children.
IVF in-vitro fertilization, may decrease the chance that two people with the sickle cell trait will pass the disease down genetically. In this type of in vitro fertilization, eggs and sperm from the parents are combined outside the womb and genetically tested for sickle cell anemia. Healthy fertilized eggs are then implanted into the mother.
Preventing Complications
Complications from sickle cell disease can include gallstones, lung crises (acute chest syndrome), pulmonary hypertension, stroke, leg ulcers that don't heal, and eye damage.
Preventing Infection
Bacterial infections can be a major complication of sickle cell disease, but often they can be prevented or treated. If a child who has sickle cell disease shows early signs of an infection, such as a fever, difficulty breathing, or localized bone pain, treatment should be given right away.
To prevent infections in babies and young children, treatments include:
Daily doses of penicillin: Treatment may begin as early as 2 months of age and continue until the child is at least 5 years old.
All routine vaccinations (including a yearly flu shot), plus vaccination(s) against streptococcus pneumonia.
Adults who have sickle cell disease should also receive flu shots every year and get vaccinated against pneumococcal infections. Both adults and children are at risk for a variety of infections, such as pneumonia and bone infections. They should be examined whenever they experience fevers, since early diagnosis and treatment result in better outcomes.
Preventing Pain
Those with more severe sickle cell anemia may benefit from daily administration of a medicine called hydroxyurea. This medicine may help reduce the number of painful crises. Hydroxyurea is used to prevent painful crises, not to treat them when they occur.
How else is a sickle cell crisis treated?
- A blood transfusion replaces blood with RBCs that are not sickle shaped.
- IV fluids treat dehydration and help reduce sickling of RBCs.
- Oxygen helps increase oxygen levels in your blood and make it easier for you to breathe.
- Surgery may be done to remove part of your spleen.
Treatment for Sickle cell
The goals of treating sickle cell disease are to prevent or relieve pain; prevent infections, organ damage, and strokes; treat anemia; and control complications.
Some doctors and clinics specialize in treating people who have sickle cell disease. Hematologists specialize in treating adults and children who have blood diseases and disorders.
Treating Pain
Mild pain is often treated with over-the-counter medicine and heating pads. Severe pain may need to be treated in a hospital. The usual treatments for acute (short-term) pain crises are fluids and pain-controlling medicines. Fluids help prevent dehydration, a condition in which the body doesn't have enough fluids. Fluids are given either by mouth or through a vein.
Common medicines used to treat pain crises include acetaminophen, nonsteroidal anti-inflammatory drugs (NSAIDs), and narcotics. Treatment for mild to moderate pain usually begins with NSAIDs or acetaminophen. If pain continues, a narcotic may be needed. Moderate to severe pain is often treated with narcotics. The narcotic may be used alone or with NSAIDs or acetaminophen.
Blood transfusions are commonly used to treat worsening anemia and sickle cell complications. Most patients with sickle cell disease have at least occasional blood transfusions. Patients with severe complications–such as stroke and acute chest syndrome–may require months or years of regular transfusions every three to four weeks to prevent ongoing damage.
Hydroxyurea treatment may be helpful in reducing crises and the need for transfusions.
People with sickle cell disease should have regular checkups to detect eye damage. And a simple ultrasound test of the head can identify children at high risk for strokes.
Sickle cell Home Remedies/Home Cure
- Fight back with fish oil
Reduce the frequency of severe pain episodes by taking a daily fish oil supplement providing 33 mg of EPA and 23 mg of DHA for every 2.2 pounds of body weight
- Fill up on fluids
Drink plenty of water and other fluids to maintain good circulation
- Take a test
Have your blood homocysteine levels checked to find out if daily folic acid supplements of 1 to 4 mg are right for you, or to discover if you have a vitamin B12 deficiency that requires treatment.
- Think zinc
Under the supervision of a doctor, take a daily supplement of 100 mg of zinc, plus 2 mg of copper, to help prevent cell damage and speed healing of leg ulcers associated with sickle cell anemia
Complications of Sickle Cell Anemia
Sickle cell disease can block the flow of blood in arteries in many parts of the body, causing many complications. The hallmark of sickle cell disease is the sickle cell crisis, which causes sudden attacks of severe pain. Acute chest syndrome, which is triggered by an infection or by blockage of blood vessels in the lungs, is another common and serious occurrence. Additional medical complications include:
- Anemia
- Bone and joint problems
- Eye damage in the retina
- Gallbladder disease
- Infections
- Kidney problems
- Leg sores and ulcers
- Liver problems
- Priapism (prolonged and painful erections)
- Pulmonary hypertension (increased pressure in the arteries of the lungs)
- Spleen damage
- Stroke





